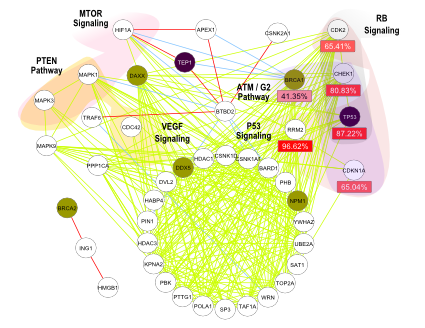
Copy Number Aberration (CNA) is a hallmark of cancer and Gene Expression (GE) changes are likely to be driven by CNA. Several studies i.e. TCGA research on ovarian cancer showed that genetic alterations and gene expression changes occurs simultaneously in RB signalling pathways. Therefore, the integration of CNA and GE data sets is of great importance specially at module-level rather than gene-level.
Such integrated data-driven information can be further filtered with protein-protein interaction (PPI) information since, it is a well known fact that, PPI hub genes are more likely to be related with cancer progression and malignance. Thus, a framework which integrates pair-wise information of CNA, GE, and PPI for cancer module identification can be of great importance in order to explain complex relationships among genes in cancer development.
A microRNA plays a key role in the development of many cancers. It is known that a microRNA regulates multiple genes and a gene is also regulated by multiple microRNAs. It is also observed that the regulation of genes by microRNAs can be different depending on patients even though they suffer from the same cancer. Hence, it is an important issue to identify these complex relationships between genes and microRNAs in cancer. Our challenge is to identify modules representing interrelationship between genes and microRNAs by integrating these data sets.
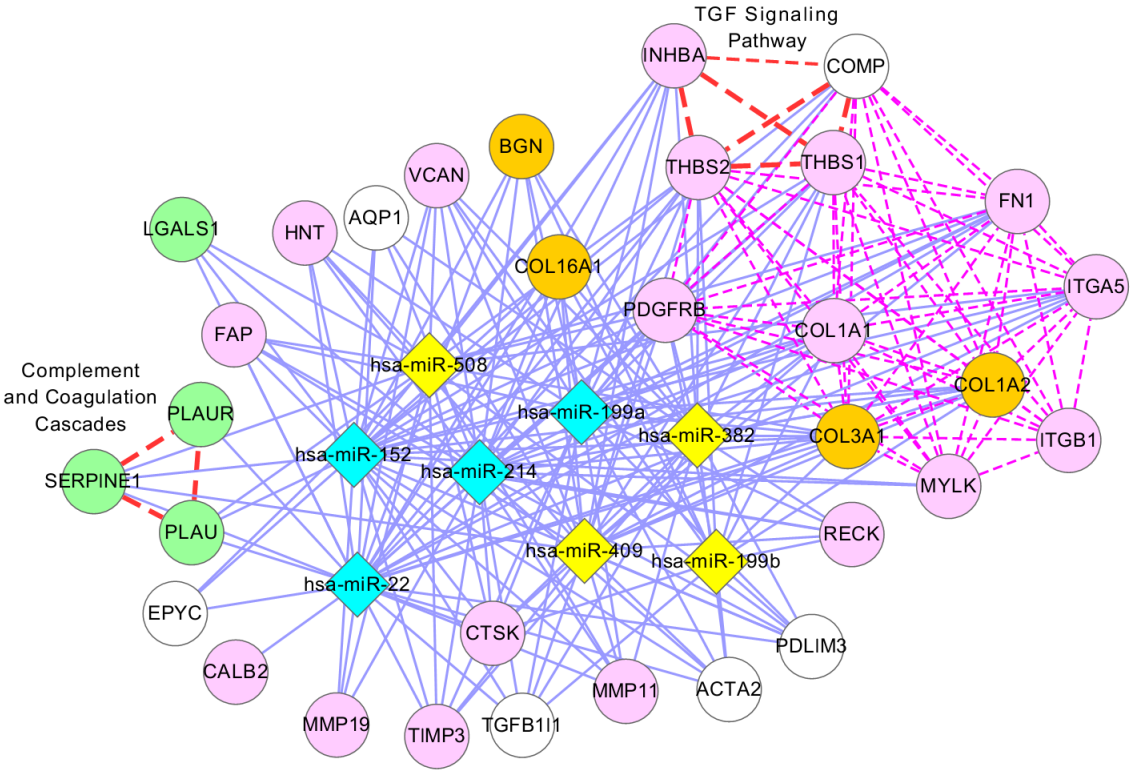
Identification of differentially expressed sub-networks from gene expression data sets has become increasingly important to our global understanding of the molecular mechanisms that drive cancer. Sub-networks can reveal the complex patterns of the whole bio-molecular network by extracting the interactions that depend on temporal or condition specific context. The identification of condition specific sub-networks is of great importance for investigating how a living cell adapts to changing environments. Our main goal is to obtain the most density sub-graph, which is described by its maximal scoring sub-network, from a whole gene regulatory network.
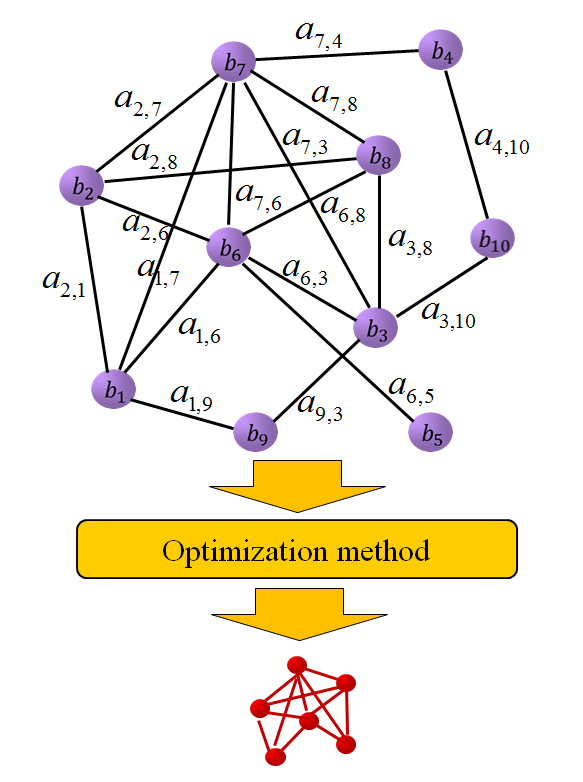
Finding maximal scoring sub-network is generally formulated as a combinatorial optimization problem. Bio-molecular networks are often large in scale. It is impossible to solve such a large combinatorial optimization problem exactly in reasonable time. We construct gene regulatory networks using some statistic based technique from gene expression datasets, and then reformulate the problems as optimization models that have strong theoretical validations.
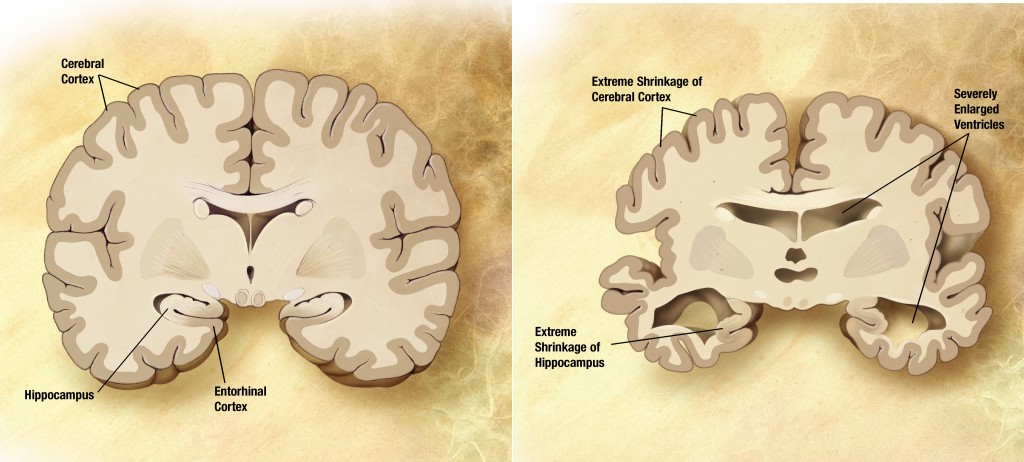
In the elderly population, Alzheimer’s disease(AD) is the main cause of dementia. It is neurodegenerative disorder leading to memory, cognitive and functional impairments and finally to death and is one of the most costly diseases for society.Since AD has complex etiology and is known to be affected by genetic components, discovery of genetic risk factors are the important to understanding the disease and developing the more effective treatment.
Although the best known genetic risk factor is the mutation in the APOE gene that increase the risk of the disease by multiple times, it does not always increase the risk. There might be large portion of the missing heritability in AD.
Our research goal is to find more genetic factors affecting onset and progression of AD using various kinds of data from genetic markers, other bio-markers such as imaging data(MRI and PET), cerebrospinal fluid(CSF) and neuropsychological assessments.
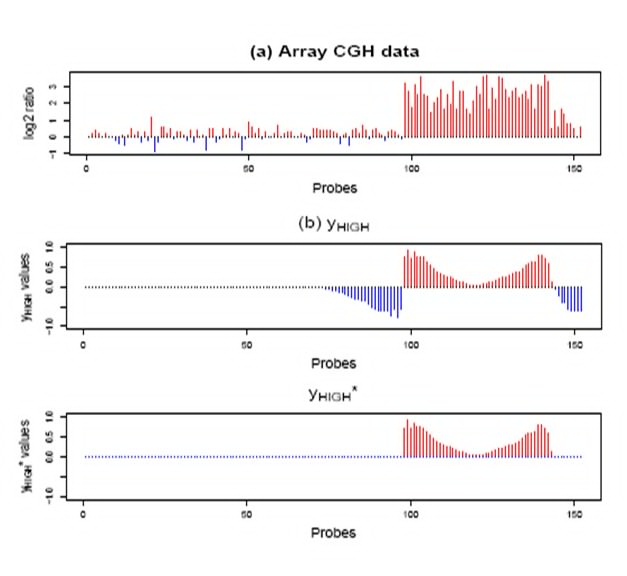
Copy number aberrations (CNAs) play important roles in cancer development, but a wide range of aberration makes it hard to distinguish key driving genes from the other neighboring genes.
Until now, with the various types of microarray data sets such as array comparative genomic hybridization (CGH) and single nucleotide polymorphism (SNP) microarrays, several methods were developed to identify cancer-driving genes.
Since next generation sequencing (NGS) data from several cancer data sets are currently available, there is a growing chance that more accurate focal aberration regions might be detected.We currently study wavelet based methods to identify aberration regions related to various types of cancers using NGS data.
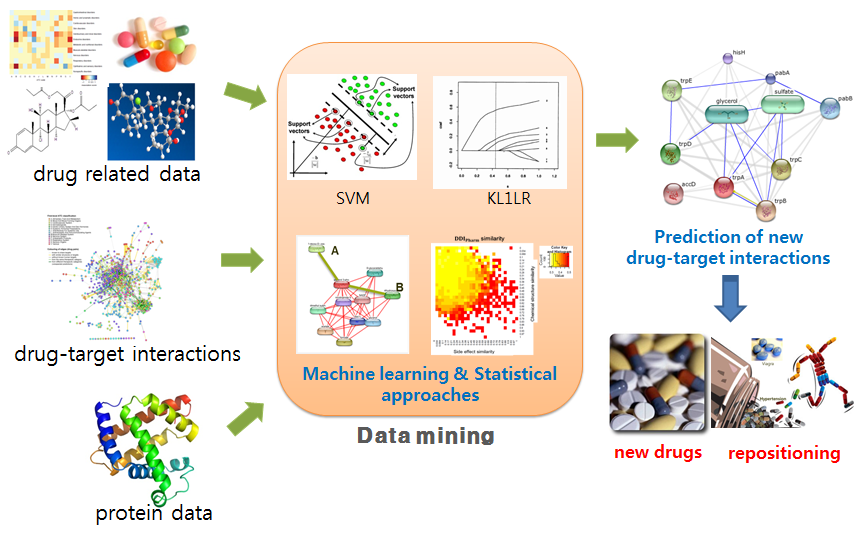
Computational methods for predicting drug-target interactions have become important in drug research because they can help to reduce the time, cost, and failure rates for developing new drugs. Recently, with the accumulation of drug-related data sets related to drug side effects and pharmacological data, it has became possible to predict potential drug-target interactions. In this study, we focus on drug-drug interactions (DDI), their adverse effect DDIAE and pharmacological information DDIPharm, and investigate the relationship among chemical structures, side effects, and DDIs from several data sources.
In this study, DDI pharm data from the STITCH database, DDIAE from drugs.com, and drug-target pairs from ChEMBL and SIDER were first collected. Then, by applying two machine learning approaches, a support vector machine (SVM) and a kernel-based L1-norm regularized logistic regression (KL1LR), we showed that DDI is a promising feature in predicting drug-target interactions. Next, the accuracies of predicting drug-target interactions using DDI were compared to those obtained using the chemical structure and side effects based on the SVM and KL1LR approaches, showing that DDI was the data source contributing the most for predicting drug-target interactions.
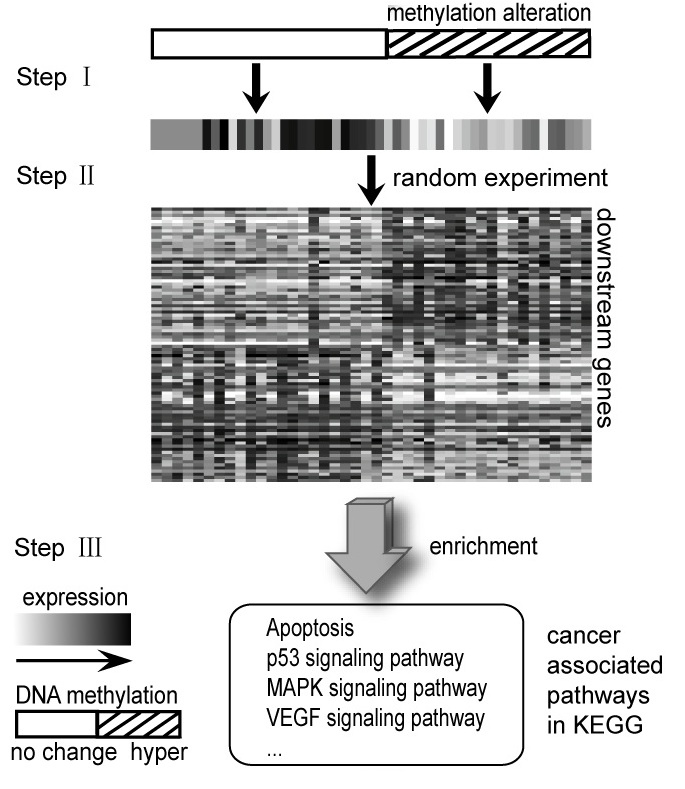
Cancer is steadily researched for many people because one of the most dangerous diseases is a cancer. These days cancer is curable if it is treated in the bud. Researchers tend to focus on early finding in cancer. There are many area to find caner occurrence. One of those is epigenetics. For example, DNA methylation regulates gene expression. It mainly occurs at CPG islands. Inhibiting gene expression of tumor suppressor is caused to hypermethylation. We study deep into this mechanism with gene expression to find caner in early stage. Two groups by DNA methylation status classify how genes express for each gene. So we could have significant genes including information between DNA methylation and gene expression.
Until now, there are a large number of methodologies to apply DNA methylation and gene experssion. Initially 5-azacytidine Experiments(McGhee&Ginder, 1979) trigger this area and then not only caner, but also various diseases are used to utilize DM data(Movassagh M et al., 2010).
To find improved methods, we read papers about relation with copy number (CN) and GE (TCGA Research Network, 2008). We tried to apply one of methods in this paper (Elizabeth Hyman et al., 2002). But it was hard to find threshold that find whether status is hypermethylation or not. We found two ways to find threshold. One of them is epigenetic variable outliers for risk prediction analysis (Andrew E. Teschendorff et al., 2012). In this paper, we used 77 normal cancer samples and 286 tumor samples to obtain threshold dividing DNA methylation status. As a result of this analysis, we could divide two groups. Then, we could get weight that indicates which gene is important. Also, among the genes, we only had differential expressed genes using t-test with normal samples.
We apply genes gotten in above to two types of methods. First, we found functions related with cancer using pathway enrichment test. Also, we performed hypergeometric test to find relation with genes-samples module that made from information between a subset of genes and a subset of samples. To summarize, through relation with information between DNA methylation and GE, we tried to find candidate cancer genes and pathway enriched about cancer. Also, we checked our genes works significantly by genes-samples module.
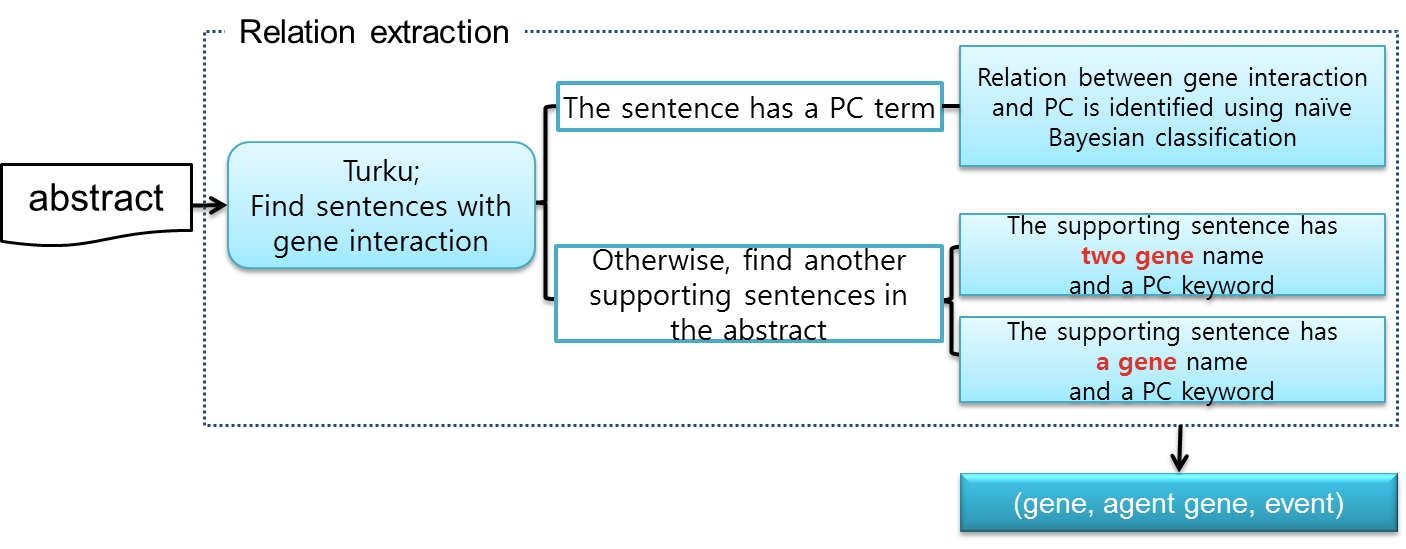
We propose a relation extraction step to find the relation between GGI and cancer. In the event extraction step, we use Turku event extraction system to nd interaction between genes. We nd a candidate sentence that has gene and event by Turku system. Then, we find an agent gene, a kind of activator or suppressor of gene expression, in the sentence parsing tree. To check that candidate sentence has relation between cancer and gene interaction, we first check that the evidence sentence has prostate cancer terms.
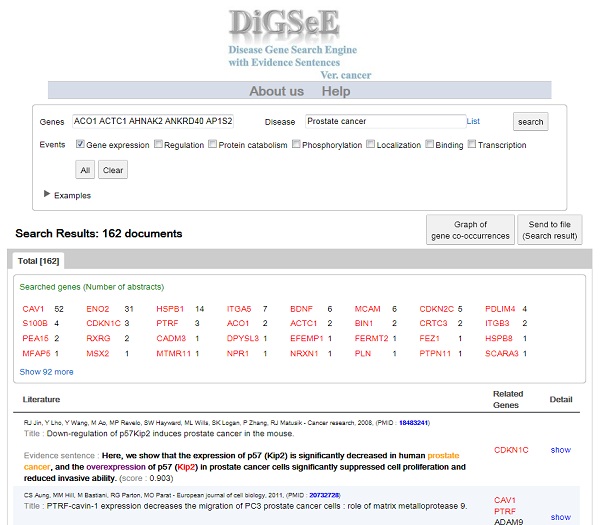
Biological events such as gene expression, regulation, phosphorylation, localization, and protein catabolism play important roles in the development of diseases. Understanding the association between diseases and genes can be enhanced with the identification of involved biological events in this association. Although biological knowledge has been accumulated in several databases and can be accessed through the Web, there is no specialized Web tool yet allowing for a query into the relationship among diseases, genes, and molecular events.
For this, We construct a disease-oriented search engine to support disease-related candidate genes from biological data sets by developing the text mining system.
